Skip to content
Understanding APBD
APBD 101
Signs and Symptoms
Undiagnosed?
An Adult-Onset Form of GSD IV
Multiple Sclerosis… or APBD?
Testing Info
Undiagnosed?
FAQs
About Us
Our Story
Timeline
Board Members/ Staff/ Volunteers
Scientific and Medical Advisory Board
Press
News Releases
APBD in the media
Legislative News
Blog
Contact us
Annual Reports
Career Opportunities
Living with APBD
Participate in Research
Patient Registry Information
Columbia University APBD Registry (CAP)
APBDRF and NORD Natural History Study (FAN)
Personal Narratives
APBD Patient Chats
Family/Caregiver Chat Events
Doctor Database
Special Disability Aids
Research
Our Research Team
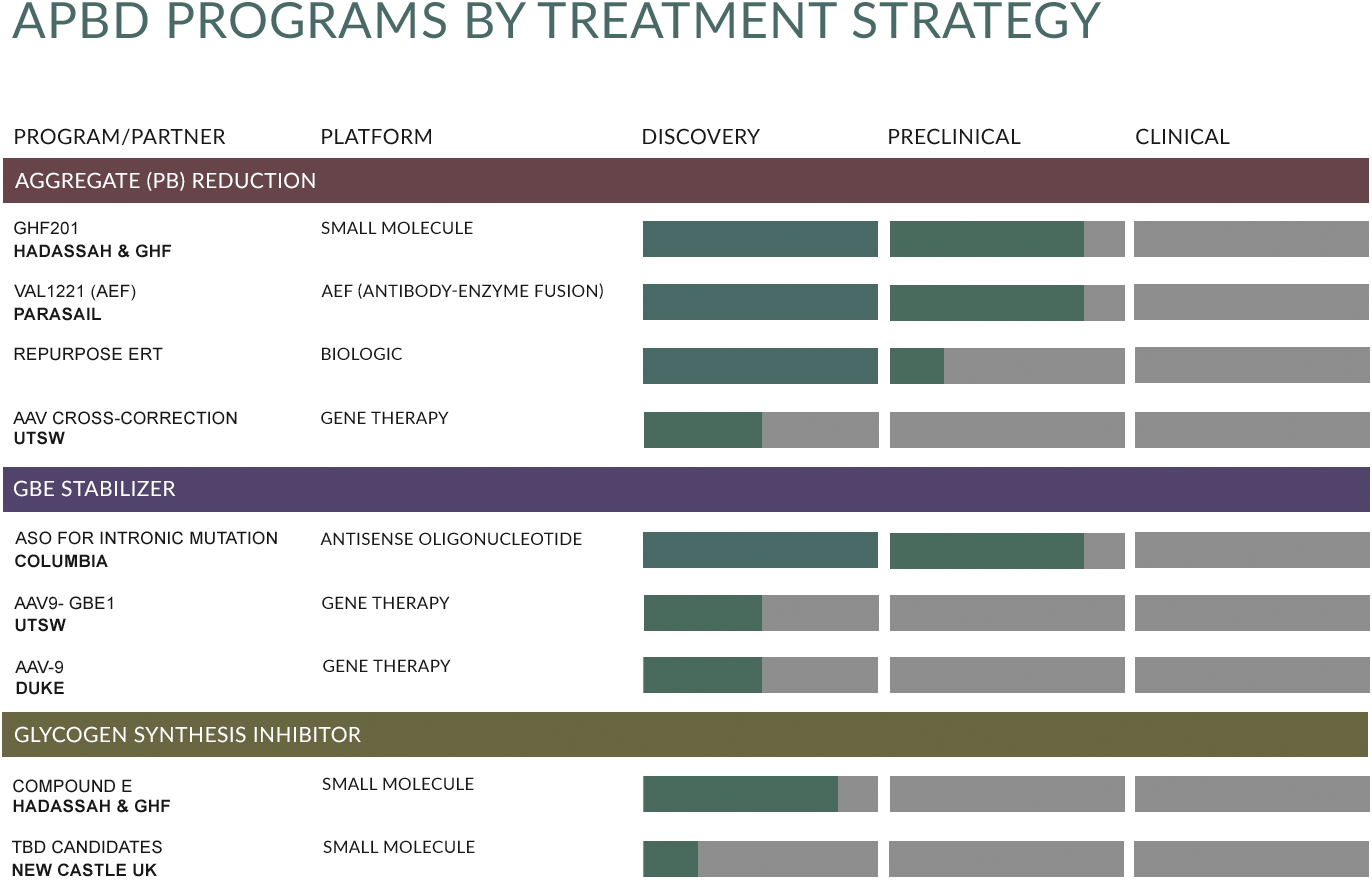
What We’re Working On
Research Highlights
Webinars
Publications
Clinical Trial Considerations
Resources
No-Cost Genetic Testing
Glossary
Videos
Links
Tool Boxes
Articles of Interest
Powerpoints
Newsletter Archive
Brochures
APBD and GSD IV Guideline
Get involved
Volunteer
APBD Planned Giving Program
Support Your #1 Cause Through Your IRA
Donate
Registry
Search for:
Search for:
What We’re Working On
Updated July 2022
Page load link
Go to Top